Incidence
Esthesioneuroblastoma (also termed olfactory neuroblastoma) is a very rare small round cell tumor arising from the nasal neuroepithelium. Less than 10% of cases occur in children and adolescents.[1,2] The estimated incidence of esthesioneuroblastoma is 0.1 cases per 100,000 people per year in children younger than 15 years.[3] In the pediatric population, the median age is 10 years, and there are no gender or racial predilections.[2]
Despite its rarity, esthesioneuroblastoma is the most common cancer of the nasal cavity in pediatric patients, accounting for 28% of cases in a Surveillance, Epidemiology, and End Results (SEER) Program study.[1]
References:
-
Benoit MM, Bhattacharyya N, Faquin W, et al.: Cancer of the nasal cavity in the pediatric population. Pediatrics 121 (1): e141-5, 2008.
-
Berger MH, Lehrich BM, Yasaka TM, et al.: Characteristics and overall survival in pediatric versus adult esthesioneuroblastoma: A population-based study. Int J Pediatr Otorhinolaryngol 144: 110696, 2021.
-
Bisogno G, Soloni P, Conte M, et al.: Esthesioneuroblastoma in pediatric and adolescent age. A report from the TREP project in cooperation with the Italian Neuroblastoma and Soft Tissue Sarcoma Committees. BMC Cancer 12: 117, 2012.
Anatomy
Figure 1 depicts the areas of the body where esthesioneuroblastoma tumors may form, including the olfactory nerve endings, olfactory bulb, nasal cavity, nasal sinuses, and brain.
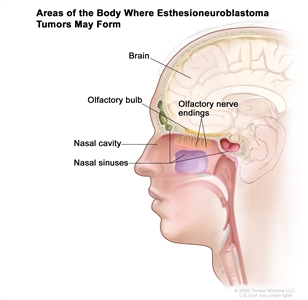
Figure 1. Esthesioneuroblastomas form in the olfactory nerve endings in the upper part of the nasal cavity. The olfactory nerves (sense of smell) pass through the many tiny holes in the bone at the base of the brain to the olfactory bulb. Esthesioneuroblastomas may spread from the nasal cavity to the nasal sinuses or to nearby tissue. They may also spread to the brain or to other parts of the body (not shown).
Clinical Presentation
Most children present with symptoms that include the following:[1]
- Nasal obstruction.
- Epistaxis.
- Hyposmia.
- Exophthalmos.
- Nasopharyngeal mass, which may have local extension into the orbits, sinuses, or frontal lobe.
References:
-
Venkatramani R, Pan H, Furman WL, et al.: Multimodality Treatment of Pediatric Esthesioneuroblastoma. Pediatr Blood Cancer 63 (3): 465-70, 2016.
Histology and Molecular Features
Esthesioneuroblastoma can be histologically confused with other small round cell tumors of the nasal cavity, including sinonasal undifferentiated carcinoma, small cell carcinoma, melanoma, and rhabdomyosarcoma. Esthesioneuroblastoma typically shows diffuse staining with neuron-specific enolase, synaptophysin, and chromogranins, with variable cytokeratin expression.[1]
Nine medical centers obtained 66 samples of olfactory neuroblastoma and tumor samples from other cancers, including alveolar rhabdomyosarcoma and sinonasal adenocarcinoma. The tumor samples were analyzed by genome-wide DNA methylation profiling, copy number analysis, immunohistochemistry, and next-generation panel sequencing. Unsupervised hierarchal clustering analysis of DNA methylation data identified the following four distinct clusters:[2]
- The largest cluster, which comprised 64% of the samples, had classical histological features of olfactory neuroblastoma. Ten percent of the cases had recurrent DNMT3A and TP53 mutations.
- A second cluster consisted of seven cases with a hypermethylator phenotype and IDH2 mutations that clustered with the group of IDH2 sinonasal carcinomas.
- A small third cluster was characterized by hypermethylation without IDH2 mutations. This result suggests that this cluster may represent a subgroup of olfactory neuroblastomas or an undefined sinonasal tumor entity.
- The fourth cluster represented a heterogenous group of 13 tumors that grouped with other entities such as sinonasal adenocarcinoma, sinonasal squamous cell carcinoma, sinonasal neuroendocrine carcinoma, and sinonasal undifferentiated carcinoma.
Using this information, the authors developed an algorithm that incorporates methylation analysis to improve the diagnostic accuracy of this entity.[2]
References:
-
Su SY, Bell D, Hanna EY: Esthesioneuroblastoma, neuroendocrine carcinoma, and sinonasal undifferentiated carcinoma: differentiation in diagnosis and treatment. Int Arch Otorhinolaryngol 18 (Suppl 2): S149-56, 2014.
-
Capper D, Engel NW, Stichel D, et al.: DNA methylation-based reclassification of olfactory neuroblastoma. Acta Neuropathol 136 (2): 255-271, 2018.
Prognostic Factors
Review of multiple case series of mainly adult patients indicates that the following may correlate with adverse prognosis:[1,2,3]
- Higher histopathological grade.
- Positive surgical margin status.
- Metastases to the cervical lymph nodes.
References:
-
Dulguerov P, Allal AS, Calcaterra TC: Esthesioneuroblastoma: a meta-analysis and review. Lancet Oncol 2 (11): 683-90, 2001.
-
Patel SG, Singh B, Stambuk HE, et al.: Craniofacial surgery for esthesioneuroblastoma: report of an international collaborative study. J Neurol Surg B Skull Base 73 (3): 208-20, 2012.
-
Herr MW, Sethi RK, Meier JC, et al.: Esthesioneuroblastoma: an update on the massachusetts eye and ear infirmary and massachusetts general hospital experience with craniofacial resection, proton beam radiation, and chemotherapy. J Neurol Surg B Skull Base 75 (1): 58-64, 2014.
Stage Information for Childhood Esthesioneuroblastoma
Tumors are staged according to the Kadish system (see Table 1). Correlated with Kadish stage, survival rates range from 90% (stage A) to less than 40% (stage D). Most patients present with locally advanced–stage disease (Kadish stages B and C), and almost one-third of patients have tumors at distant sites (Kadish stage D).[1,2,3,4]
Recent reports suggest that positron emission tomography–computed tomography (PET-CT) may aid in staging the disease.[5]
Table 1. Kadish Staging System
| Stage |
Description |
| A |
Tumor confined to the nasal cavity. |
| B |
Tumor extending to the nasal sinuses. |
| C |
Tumor extending to the nasal sinuses and beyond. |
| D |
Tumor metastases present. |
References:
-
Bisogno G, Soloni P, Conte M, et al.: Esthesioneuroblastoma in pediatric and adolescent age. A report from the TREP project in cooperation with the Italian Neuroblastoma and Soft Tissue Sarcoma Committees. BMC Cancer 12: 117, 2012.
-
Benoit MM, Bhattacharyya N, Faquin W, et al.: Cancer of the nasal cavity in the pediatric population. Pediatrics 121 (1): e141-5, 2008.
-
Venkatramani R, Pan H, Furman WL, et al.: Multimodality Treatment of Pediatric Esthesioneuroblastoma. Pediatr Blood Cancer 63 (3): 465-70, 2016.
-
Berger MH, Lehrich BM, Yasaka TM, et al.: Characteristics and overall survival in pediatric versus adult esthesioneuroblastoma: A population-based study. Int J Pediatr Otorhinolaryngol 144: 110696, 2021.
-
Broski SM, Hunt CH, Johnson GB, et al.: The added value of 18F-FDG PET/CT for evaluation of patients with esthesioneuroblastoma. J Nucl Med 53 (8): 1200-6, 2012.
Special Considerations for the Treatment of Children With Cancer
Cancer in children and adolescents is rare, although the overall incidence has been slowly increasing since 1975.[1] Referral to medical centers with multidisciplinary teams of cancer specialists experienced in treating cancers that occur in childhood and adolescence should be considered. This multidisciplinary team approach incorporates the skills of the following health care professionals and others to ensure that children receive treatment, supportive care, and rehabilitation that will achieve optimal survival and quality of life:
- Primary care physicians.
- Pediatric surgeons.
- Radiation oncologists.
- Pediatric medical oncologists/hematologists.
- Rehabilitation specialists.
- Pediatric nurse specialists.
- Social workers.
- Child-life professionals.
- Psychologists.
For information about supportive care for children and adolescents with cancer, see the summaries on Supportive and Palliative Care.
The American Academy of Pediatrics has outlined guidelines for pediatric cancer centers and their role in the treatment of pediatric patients with cancer.[2] At these pediatric cancer centers, clinical trials are available for most types of cancer that occur in children and adolescents, and the opportunity to participate is offered to most patients and their families. Clinical trials for children and adolescents diagnosed with cancer are generally designed to compare potentially better therapy with current standard therapy. Most of the progress made in identifying curative therapy for childhood cancers has been achieved through clinical trials. Information about ongoing clinical trials is available from the NCI website.
Dramatic improvements in survival have been achieved for children and adolescents with cancer. Between 1975 and 2020, childhood cancer mortality decreased by more than 50%.[3,4,5] Childhood and adolescent cancer survivors require close monitoring because side effects of cancer therapy may persist or develop months or years after treatment. For information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors, see Late Effects of Treatment for Childhood Cancer.
Childhood cancer is a rare disease, with about 15,000 cases diagnosed annually in the United States in individuals younger than 20 years.[6] The U.S. Rare Diseases Act of 2002 defines a rare disease as one that affects populations smaller than 200,000 people. Therefore, all pediatric cancers are considered rare.
The designation of a rare tumor is not uniform among pediatric and adult groups. In adults, rare cancers are defined as those with an annual incidence of fewer than six cases per 100,000 people. They account for up to 24% of all cancers diagnosed in the European Union and about 20% of all cancers diagnosed in the United States.[7,8] Also, the designation of a pediatric rare tumor is not uniform among international groups, as follows:
These rare cancers are extremely challenging to study because of the low number of patients with any individual diagnosis, the predominance of rare cancers in the adolescent population, and the lack of clinical trials for adolescents with rare cancers.
References:
-
Smith MA, Seibel NL, Altekruse SF, et al.: Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 28 (15): 2625-34, 2010.
-
American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed December 8, 2022.
-
Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
-
National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 6, 2022.
-
Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed September 7, 2022.
-
Ward E, DeSantis C, Robbins A, et al.: Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin 64 (2): 83-103, 2014 Mar-Apr.
-
Gatta G, Capocaccia R, Botta L, et al.: Burden and centralised treatment in Europe of rare tumours: results of RARECAREnet-a population-based study. Lancet Oncol 18 (8): 1022-1039, 2017.
-
DeSantis CE, Kramer JL, Jemal A: The burden of rare cancers in the United States. CA Cancer J Clin 67 (4): 261-272, 2017.
-
Ferrari A, Brecht IB, Gatta G, et al.: Defining and listing very rare cancers of paediatric age: consensus of the Joint Action on Rare Cancers in cooperation with the European Cooperative Study Group for Pediatric Rare Tumors. Eur J Cancer 110: 120-126, 2019.
-
Pappo AS, Krailo M, Chen Z, et al.: Infrequent tumor initiative of the Children's Oncology Group: initial lessons learned and their impact on future plans. J Clin Oncol 28 (33): 5011-6, 2010.
Treatment of Childhood Esthesioneuroblastoma
The use of multimodal therapy optimizes the chances for survival, with more than 70% of children expected to survive 5 or more years after initial diagnosis.[1,2,3,4,5] Neuromeningeal progression is the most common type of treatment failure.[5,6][Level of evidence C1]
Treatment options according to Kadish stage include the following:[7]
-
Kadish stage A: Surgery alone with clear margins. Adjuvant radiation therapy is indicated in patients with close and positive margins or with residual disease.
-
Kadish stage B: Surgery followed by adjuvant radiation therapy. The role of adjuvant chemotherapy is controversial.
-
Kadish stage C: Neoadjuvant approach with chemotherapy, radiation therapy, or concurrent chemotherapy-radiation therapy followed by surgery.
-
Kadish stage D: Systemic chemotherapy and radiation therapy to local and metastatic sites.
The mainstay of treatment is surgery and radiation therapy. However, esthesioneuroblastoma is a chemosensitive neoplasm, and the use of neoadjuvant chemotherapy can facilitate resection.[5,7,8] Endoscopic sinus surgery offers short-term outcomes similar to open craniofacial resection.[9]; [10][Level of evidence C2] Other techniques such as stereotactic radiosurgery and proton-beam therapy (charged-particle radiation therapy) may also play a role in the management of this tumor.[3,11]
Nodal metastases are seen in about 5% of patients. Routine neck dissection and nodal exploration are not indicated in the absence of clinical or radiological evidence of disease.[12] Management of cervical lymph node metastases has been addressed in a review article.[12]
Reports indicate promising results with the increased use of resection and neoadjuvant or adjuvant chemotherapy in patients with advanced-stage disease.[2,5,13,14,15]; [16][Level of evidence C1] Chemotherapy regimens that have been used with efficacy include the following:
- Cisplatin and etoposide with or without ifosfamide.[17,18]
- Vincristine, dactinomycin, and cyclophosphamide with or without doxorubicin.
- Ifosfamide and etoposide.
- Cisplatin plus etoposide or doxorubicin.[2]
- Vincristine, doxorubicin, and cyclophosphamide.[19]
- Irinotecan plus docetaxel.[20][Level of evidence C1]
References:
-
Bisogno G, Soloni P, Conte M, et al.: Esthesioneuroblastoma in pediatric and adolescent age. A report from the TREP project in cooperation with the Italian Neuroblastoma and Soft Tissue Sarcoma Committees. BMC Cancer 12: 117, 2012.
-
Eich HT, Müller RP, Micke O, et al.: Esthesioneuroblastoma in childhood and adolescence. Better prognosis with multimodal treatment? Strahlenther Onkol 181 (6): 378-84, 2005.
-
Lucas JT, Ladra MM, MacDonald SM, et al.: Proton therapy for pediatric and adolescent esthesioneuroblastoma. Pediatr Blood Cancer 62 (9): 1523-8, 2015.
-
Berger MH, Lehrich BM, Yasaka TM, et al.: Characteristics and overall survival in pediatric versus adult esthesioneuroblastoma: A population-based study. Int J Pediatr Otorhinolaryngol 144: 110696, 2021.
-
Venkatramani R, Pan H, Furman WL, et al.: Multimodality Treatment of Pediatric Esthesioneuroblastoma. Pediatr Blood Cancer 63 (3): 465-70, 2016.
-
Dumont B, Fresneau B, Claude L, et al.: Pattern of loco-regional relapses and treatment in pediatric esthesioneuroblastoma: The French very rare tumors group (Fracture) contribution. Pediatr Blood Cancer 67 (4): e28154, 2020.
-
Safi C, Spielman D, Otten M, et al.: Treatment Strategies and Outcomes of Pediatric Esthesioneuroblastoma: A Systematic Review. Front Oncol 10: 1247, 2020.
-
Ozsahin M, Gruber G, Olszyk O, et al.: Outcome and prognostic factors in olfactory neuroblastoma: a rare cancer network study. Int J Radiat Oncol Biol Phys 78 (4): 992-7, 2010.
-
Soler ZM, Smith TL: Endoscopic versus open craniofacial resection of esthesioneuroblastoma: what is the evidence? Laryngoscope 122 (2): 244-5, 2012.
-
Gallia GL, Reh DD, Lane AP, et al.: Endoscopic resection of esthesioneuroblastoma. J Clin Neurosci 19 (11): 1478-82, 2012.
-
Unger F, Haselsberger K, Walch C, et al.: Combined endoscopic surgery and radiosurgery as treatment modality for olfactory neuroblastoma (esthesioneuroblastoma). Acta Neurochir (Wien) 147 (6): 595-601; discussion 601-2, 2005.
-
Zanation AM, Ferlito A, Rinaldo A, et al.: When, how and why to treat the neck in patients with esthesioneuroblastoma: a review. Eur Arch Otorhinolaryngol 267 (11): 1667-71, 2010.
-
Kumar M, Fallon RJ, Hill JS, et al.: Esthesioneuroblastoma in children. J Pediatr Hematol Oncol 24 (6): 482-7, 2002 Aug-Sep.
-
Loy AH, Reibel JF, Read PW, et al.: Esthesioneuroblastoma: continued follow-up of a single institution's experience. Arch Otolaryngol Head Neck Surg 132 (2): 134-8, 2006.
-
Porter AB, Bernold DM, Giannini C, et al.: Retrospective review of adjuvant chemotherapy for esthesioneuroblastoma. J Neurooncol 90 (2): 201-4, 2008.
-
Benfari G, Fusconi M, Ciofalo A, et al.: Radiotherapy alone for local tumour control in esthesioneuroblastoma. Acta Otorhinolaryngol Ital 28 (6): 292-7, 2008.
-
Kim DW, Jo YH, Kim JH, et al.: Neoadjuvant etoposide, ifosfamide, and cisplatin for the treatment of olfactory neuroblastoma. Cancer 101 (10): 2257-60, 2004.
-
Kumar R: Esthesioneuroblastoma: Multimodal management and review of literature. World J Clin Cases 3 (9): 774-8, 2015.
-
El Kababri M, Habrand JL, Valteau-Couanet D, et al.: Esthesioneuroblastoma in children and adolescent: experience on 11 cases with literature review. J Pediatr Hematol Oncol 36 (2): 91-5, 2014.
-
Kiyota N, Tahara M, Fujii S, et al.: Nonplatinum-based chemotherapy with irinotecan plus docetaxel for advanced or metastatic olfactory neuroblastoma: a retrospective analysis of 12 cases. Cancer 112 (4): 885-91, 2008.
Treatment Options Under Clinical Evaluation for Childhood Esthesioneuroblastoma
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
Changes to This Summary (12 / 09 / 2022)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Special Considerations for the Treatment of Children With Cancer
Revised text to state that between 1975 and 2020, childhood cancer mortality decreased by more than 50% (cited National Cancer Institute as reference 4 and Surveillance Research Program, National Cancer Institute as reference 5).
Revised text to state that rare pediatric cancers account for about 5% of the cancers diagnosed in children aged 0 to 14 years and about 27% of the cancers diagnosed in adolescents aged 15 to 19 years. Also revised text to state that most cancers in subgroup XI are either melanomas or thyroid cancers, with other cancer types accounting for only 2% of the cancers in children aged 0 to 14 years and 9.3% of cancers in adolescents aged 15 to 19 years.
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of childhood esthesioneuroblastoma. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Childhood Esthesioneuroblastoma Treatment are:
- Denise Adams, MD (Children's Hospital Boston)
- Karen J. Marcus, MD, FACR (Dana-Farber Cancer Institute/Boston Children's Hospital)
- William H. Meyer, MD (University of Oklahoma Health Sciences Center)
- Paul A. Meyers, MD (Memorial Sloan-Kettering Cancer Center)
- Thomas A. Olson, MD (Aflac Cancer and Blood Disorders Center of Children's Healthcare of Atlanta - Egleston Campus)
- Alberto S. Pappo, MD (St. Jude Children's Research Hospital)
- D. Williams Parsons, MD, PhD
- Arthur Kim Ritchey, MD (Children's Hospital of Pittsburgh of UPMC)
- Carlos Rodriguez-Galindo, MD (St. Jude Children's Research Hospital)
- Stephen J. Shochat, MD (St. Jude Children's Research Hospital)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as "NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary]."
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Childhood Esthesioneuroblastoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/head-and-neck/hp/child/esthesioneuroblastoma-treatment-pdq. Accessed <MM/DD/YYYY>.
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either "standard" or "under clinical evaluation." These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website's Email Us.
Last Revised: 2022-12-09
